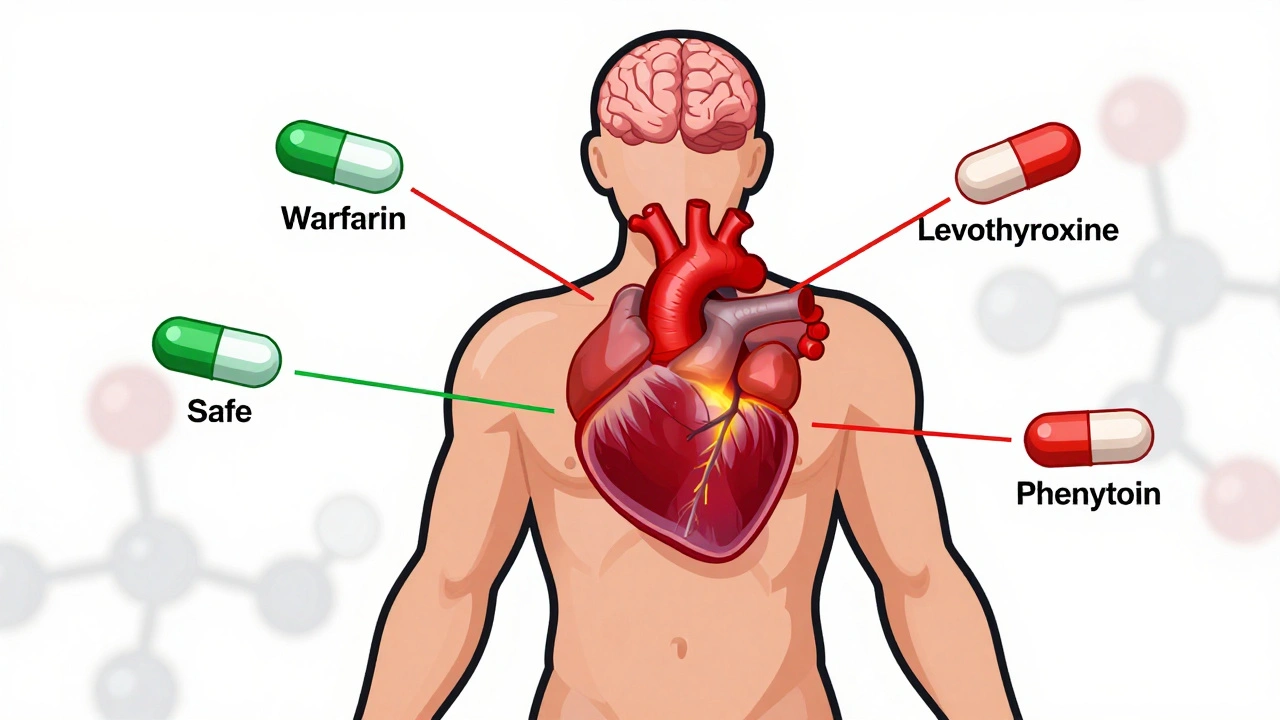
When a patient switches from a brand-name drug to a generic, most people assume it’s a simple swap-same active ingredient, same effect. But for NTI generics, that assumption can be dangerous. Narrow Therapeutic Index (NTI) drugs have razor-thin margins between a therapeutic dose and a toxic one. A 5% difference in blood concentration might mean the difference between seizure control and a life-threatening episode, or between stable blood thinning and dangerous bleeding. This isn’t theoretical. Real patients have experienced thyroid fluctuations after switching levothyroxine generics. Others have had INR levels spike after changing warfarin brands. And yet, across the globe, regulators handle these drugs in wildly different ways.
Why NTI Generics Are Different
NTI drugs like warfarin, phenytoin, digoxin, and levothyroxine aren’t just any generics. They’re high-risk medications where small changes in absorption, metabolism, or formulation can lead to serious harm. The FDA defines NTI drugs as those where small differences in dose or blood concentration may cause serious therapeutic failures or adverse reactions. That’s why the approval standards for these generics are far stricter than for regular generics.
For most generic drugs, regulators accept bioequivalence within an 80-125% range. That means the generic’s blood concentration can be 20% lower or 25% higher than the brand and still be considered equivalent. For NTI drugs, that range is often tightened. In the U.S., the FDA now requires bioequivalence limits of 90-111% for many NTI drugs-sometimes even tighter. The quality assay, which measures how much active ingredient is in each tablet, is also more precise: 95-105% instead of the standard 90-110%. These aren’t arbitrary numbers. They’re based on clinical data showing that even minor deviations can trigger adverse events.
How the U.S. Handles NTI Generics
The FDA has led the way in tightening NTI standards. Since 2010, it has issued over 100 drug-specific guidance documents for NTI products. For drugs like levothyroxine and phenytoin, the agency requires more rigorous bioequivalence studies, often using healthy volunteers instead of patients to eliminate variability from disease states. Studies must include multi-point dissolution profiles and stress testing to predict how the drug behaves over time and under different conditions.
But regulation doesn’t stop at approval. In 26 U.S. states, pharmacists can’t substitute an NTI generic without special consent. North Carolina requires written approval from both the prescriber and patient. Connecticut, Idaho, and Illinois mandate extra notifications for anti-epileptic drugs. These rules exist because pharmacists report frequent calls from doctors asking them not to substitute. A 2019 survey found 67% of U.S. pharmacists received such requests-78% of them for anti-epileptic drugs, 63% for warfarin.
Despite these safeguards, problems persist. In 2023, the FDA reported a 22% higher rejection rate for NTI generic applications compared to non-NTI ones. The main reason? Bioequivalence failures. One recalled antihypertensive generic in 2021 contained nitrosamine impurities-something that could have been caught with better testing. The cost to develop an NTI generic? $5-7 million and 18-24 months, nearly double the time and money needed for regular generics.
The European Union: Fragmented but Strict
In Europe, the regulatory picture is more complex. The European Medicines Agency (EMA) offers three approval paths: the Centralized Procedure (CP), National Procedure (NP), and Mutual Recognition. Only about 32% of NTI generics use the Centralized Procedure today, but that number has grown from 42% in 2018. The CP takes about 210 days and results in approval across all EU member states. But most companies still opt for National Procedures, which can take 12-18 months and vary by country.
Unlike the U.S., the EU doesn’t have a single set of substitution rules. Each country decides whether pharmacists can switch NTI generics. Some, like Spain, have aggressive pricing policies: the first generic must be priced at least 40% below the brand. Subsequent generics must match or undercut that price. Other countries, like Germany and the UK, allow more flexibility. This creates confusion. A 2022 survey by the European Association of Hospital Pharmacists found that 58% of pharmacists struggled to understand substitution rules across borders-especially when a drug was approved under the Decentralized Procedure in one country but not another.
Still, Europe has strong safety data. A 2021 IMS Institute study of 12,500 patients across 15 EU countries found that when strict bioequivalence standards were followed, 94.7% of NTI generic switches resulted in equivalent clinical outcomes. The key? Consistency in testing and enforcement.

Canada, Japan, and Other Regulators
Canada takes a pragmatic approach. It allows foreign-sourced reference products for bioequivalence studies if they match the original drug’s solubility, formulation, and physicochemical properties. This helps manufacturers avoid costly delays. Japan’s PMDA has detailed guidance for topical NTI drugs, recognizing that skin absorption can vary dramatically between formulations. Both countries have specific, science-driven standards-unlike many emerging markets where regulatory guidance is either lacking or inconsistent.
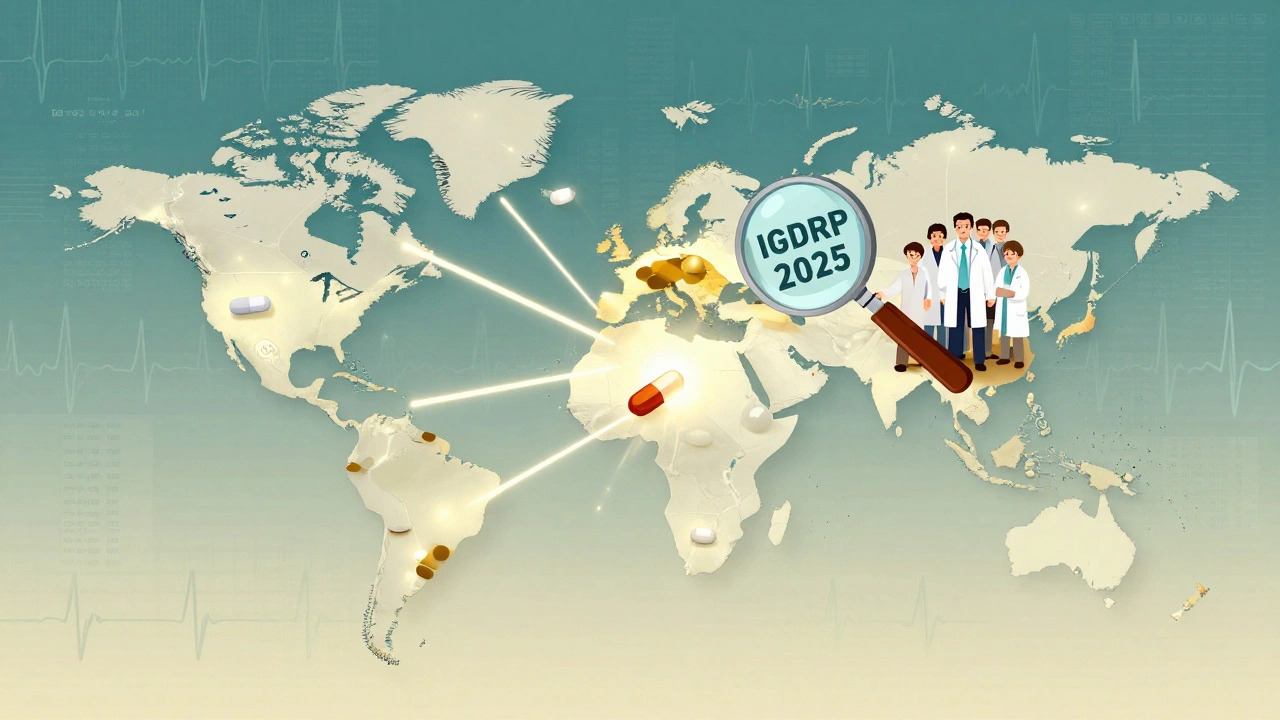
Only four major regulators-Canada, EMA, Japan, and the U.S.-have published detailed NTI-specific bioequivalence guidance. Brazil, Mexico, Singapore, and South Korea do not. This creates a global patchwork. A generic approved in the U.S. might not meet Canadian standards, and vice versa. That’s why the International Generic Drug Regulators Pilot (IGDRP), launched in 2012, exists. It includes regulators from the U.S., EU, Canada, Japan, South Korea, Singapore, Switzerland, and Taiwan. Their goal? Harmonize standards so one high-quality generic can be sold globally without retesting.
What’s Changing in 2025?
The landscape is shifting. In 2023, the ICH adopted the M9 guideline on biowaivers, which could simplify approval for some NTI drugs that meet specific solubility and permeability criteria. The FDA’s GDUFA III, also launched in 2023, includes new post-marketing surveillance requirements for NTI generics-meaning the agency will monitor real-world outcomes more closely after approval.
By 2025, the FDA plans to move from traditional average bioequivalence to population bioequivalence for certain NTI drugs. This means testing not just the average concentration across a group of volunteers, but how the drug behaves across different body types, metabolisms, and genetic profiles. It’s a more realistic way to predict how a drug will work in the real world.
Meanwhile, the EMA is pushing more companies toward the Centralized Procedure. With 68% of new generic applications now using it in 2022, the trend is clear: centralized approval reduces duplication, improves consistency, and speeds up access.
What This Means for Prescribers and Patients
If you’re prescribing an NTI drug, you can’t assume all generics are equal. Even if a generic is FDA-approved, it might not behave the same in your patient. Some patients do fine switching. Others don’t. There’s no universal rule.
Here’s what you should do:
- Know your patient’s history. Have they had issues with generic switches before?
- Check your state’s substitution laws. Some require consent; others don’t.
- When possible, stick with the same manufacturer. Even if two generics are technically bioequivalent, different formulations can behave differently over time.
- Monitor closely after a switch. For warfarin, check INR within 3-5 days. For levothyroxine, check TSH in 6-8 weeks.
Patients deserve safe, affordable access to generics. But safety can’t be sacrificed for cost. The best generics aren’t the cheapest-they’re the ones proven to work consistently, in every patient, every time.
Market Trends and Key Players
The global NTI generics market was worth $48.7 billion in 2022 and is expected to hit $72.3 billion by 2027. The U.S. leads with 42% of sales; Europe follows at 34%. Teva is the market leader with nearly 19% share, followed by Mylan, Sandoz, and Hikma. But growth isn’t easy. With rejection rates higher and development costs steeper, only the most thorough manufacturers succeed.
Warfarin generics have nearly 92% market penetration in the U.S.-patients and providers trust them. But levothyroxine? Only 67%. That gap tells you everything. Trust isn’t about regulation. It’s about experience. And when patients have had bad outcomes, they-and their doctors-hesitate.
Looking Ahead
The future of NTI generics lies in global alignment. If regulators can agree on a single set of high standards-testing methods, bioequivalence limits, post-market monitoring-then one high-quality generic can be trusted everywhere. That’s the goal of IGDRP. That’s what the FDA and EMA are moving toward. But progress is slow. Until then, prescribers must remain vigilant. Patients must be informed. And regulators must remember: when the margin between safety and danger is this thin, there’s no room for compromise.
Comments
Mark Ziegenbein
Let me be clear-this isn’t about generics. It’s about control. The FDA doesn’t care about your thyroid levels, they care about the bottom line. They’ve been letting these companies cut corners since the 90s and now they’re slapping on a 90-111% bioequivalence bandaid like it’s magic. You think that’s science? It’s accounting. I’ve seen patients go from stable to seizing because a pharmacist swapped their levothyroxine for the cheapest one on the shelf. The system is rigged. The manufacturers? They’re laughing all the way to the bank. And you? You’re just another data point in their quarterly report.
And don’t get me started on the IGDRP. That’s just a PR stunt. The same people who approved the nitrosamine-contaminated antihypertensives are now talking about harmonization. Wake up. There’s no global standard. There’s just profit disguised as policy.
Prescribers? You’re not the heroes here. You’re the enablers. You let pharmacists switch without a second thought. You don’t track INR for 3 days-you just refill the script. You want safe meds? Stop trusting the system. Start writing ‘Do Not Substitute’ on every NTI prescription. And if your state doesn’t let you? Move to a country that actually gives a damn.
December 6, 2025 at 02:37
Rupa DasGupta
I’m from India and we don’t even have proper NTI guidelines 😭 My cousin took a generic warfarin and ended up in ICU. The pharmacy just gave her the cheapest one. No tests. No warnings. Just ‘it’s the same thing’. 💔 Now she’s on blood thinners for life and her family is broke. Why does America have all these rules and we get garbage? 🤬
December 7, 2025 at 00:45
Marvin Gordon
Real talk-this is one of the most important posts I’ve read this year. I’m a pharmacist in rural Ohio and I see this every week. Patients don’t understand why their doctor won’t let them switch. They think it’s just about cost. But the truth? It’s about consistency. One guy came in with his INR at 8.2 after switching to a new generic warfarin. He almost bled out. We had to call the ER. That’s not a ‘maybe’. That’s a ‘this happened because we let it happen’.
And yeah, the system’s broken. But the solution isn’t to throw out generics. It’s to demand better. Push for the same manufacturer. Demand that your pharmacy log substitutions. Educate your patients. We’re not powerless here. We’re just lazy.
Let’s stop treating NTI drugs like aspirin. They’re not. And if we treat them like the high-risk meds they are? We can save lives. Not just paperwork.
December 8, 2025 at 08:45
ashlie perry
the fda is controlled by big pharma and they want you to think generics are safe but theyre not theyre testing on poor people in india and then selling the same stuff here for 3x the price and no one talks about this because theyre all in on it
December 9, 2025 at 06:35
Kylee Gregory
It’s interesting how we frame this as a regulatory failure, but maybe it’s a philosophical one. We’ve built a healthcare system that values efficiency over individuality. We assume bioequivalence is enough because we treat patients as averages. But people aren’t averages. A 5% variation in blood concentration doesn’t affect everyone the same way. One person’s stable is another person’s seizure.
What if the real question isn’t ‘how do we regulate generics better?’ but ‘how do we stop treating human biology like a spreadsheet?’
Maybe the answer isn’t tighter limits-it’s personalized medicine. Maybe the future isn’t more testing-it’s more listening. To the patient who says ‘I don’t feel right on this one.’ To the doctor who says ‘I’ve seen this before.’ To the pharmacist who says ‘I don’t want to swap this.’
Regulation can’t fix what we refuse to see.
December 9, 2025 at 12:05
Philip Kristy Wijaya
It is imperative to underscore that the current regulatory paradigm, while ostensibly grounded in empirical science, is in fact a product of institutional inertia and bureaucratic compromise. The FDA’s 90-111% bioequivalence window, though ostensibly more stringent than the conventional 80-125%, remains fundamentally inadequate when applied to drugs with narrow therapeutic indices. One must consider the pharmacokinetic variance inherent in polymorphic metabolizers, the influence of gut microbiota on drug absorption, and the destabilizing effects of dietary interactions-all of which are systematically excluded from bioequivalence protocols.
Moreover, the assertion that ‘one generic is as good as another’ is not merely erroneous-it is an epistemological travesty. The molecular architecture of excipients, the crystalline form of the active pharmaceutical ingredient, and the compression force during tablet manufacturing are all variables that, while statistically insignificant in population studies, are clinically catastrophic in individual cases.
Therefore, it is not sufficient to advocate for ‘better’ regulation. We must demand a paradigm shift: mandatory individualized pharmacokinetic monitoring for all NTI drug recipients, coupled with a registry of generic manufacturers and their formulation profiles. Anything less is not negligence-it is malfeasance.
December 11, 2025 at 09:19
Jennifer Patrician
They’re lying. Everyone knows the FDA approves generics from China that are made in the same factories as the brand but with cheaper filler. They don’t test them properly. They just rubber stamp it. I heard a nurse say they found a generic warfarin with actual glass particles in it. They covered it up. And now they want us to trust ‘population bioequivalence’? Like we’re lab rats? Wake up. This is a scam. The only reason they’re pushing this is because insurance companies want to save $2 a pill. Someone’s dying for that $2.
December 12, 2025 at 21:07
Mellissa Landrum
usa is the only country that cares about this. everyone else just lets pharmacists swap anything. why? because they dont care about people. we got the best doctors and the worst system. america is the only country that makes you pay for medicine to live. so yeah make the generics expensive and hard to make. because we dont want them to be cheap. we want them to be safe. and if you disagree you hate america
December 14, 2025 at 11:48