When you pick up a generic pill at the pharmacy, you might assume it’s just a cheaper version of the brand-name drug. But here’s the real question: how do we know it works the same way in your body? The answer lies in a quiet but powerful standard called the 80-125% rule. It’s not about how much active ingredient is in the pill. It’s not about price. It’s about what happens after you swallow it - how fast and how much of the drug enters your bloodstream. This rule is the invisible gatekeeper that ensures generic drugs are just as safe and effective as their brand-name counterparts.
What the 80-125% Rule Actually Means
Many people think the 80-125% rule means generic drugs can contain anywhere from 80% to 125% of the active ingredient compared to the brand-name version. That’s a common misunderstanding. In reality, the rule doesn’t refer to the amount of drug in the tablet at all. It refers to the pharmacokinetic response - how your body absorbs and processes the drug after you take it.
Regulators measure two key things: AUC (Area Under the Curve) and Cmax (maximum concentration). AUC tells you the total amount of drug your body is exposed to over time. Cmax tells you how quickly the drug reaches its peak level in your blood. These numbers are measured in clinical studies using healthy volunteers who take both the generic and the brand-name drug in a crossover design.
The 80-125% rule says that the 90% confidence interval of the ratio between the generic and brand-name drug’s geometric mean AUC and Cmax must fall entirely within 80% to 125%. That means if the brand-name drug delivers 100 units of exposure, the generic must deliver between 80 and 125 units - not as a range of possible values, but as a statistically proven result.
This isn’t arbitrary. The numbers come from logarithmic transformation of data. Pharmacokinetic data doesn’t follow a normal bell curve - it’s skewed. Taking the log of AUC and Cmax makes the distribution symmetrical, and the 80-125% range becomes a clean ±20% on that log scale. The 90% confidence interval is used because it allows for a 5% risk of error on each end - a total 10% risk of being wrong, which regulators deemed acceptable after decades of real-world evidence.
Why This Rule Exists - And Why It Works
Before the 80-125% rule, regulators used looser standards. In the 1970s, some agencies required that 75% of subjects show ratios between 75% and 133%. That wasn’t precise enough. The shift to the 80-125% rule in the 1980s was a game-changer. It was based on expert consensus, not clinical trials - but since then, real-world data has proven it right.
The FDA, EMA, WHO, and Health Canada all use this standard. It’s not a coincidence. This harmonization lets drug companies develop generics once and get approval in dozens of countries. Without it, the global generic drug market - worth over $227 billion - wouldn’t exist in its current form.
Post-market surveillance tells the real story. A 2020 FDA analysis of over 2,000 generic drugs approved between 2003 and 2016 found that only 0.34% ever needed label changes due to bioequivalence issues. That’s fewer than 1 in 300. In contrast, a 2022 survey of pharmacists showed that 63% still thought the rule meant generics could have 80-125% less active ingredient. That’s not how it works. Generic pills typically contain 95-105% of the labeled amount - same as brand-name drugs. The rule is about how your body handles it, not what’s in the pill.

When the Rule Isn’t Enough
The 80-125% rule is a one-size-fits-all standard. And that’s its biggest weakness.
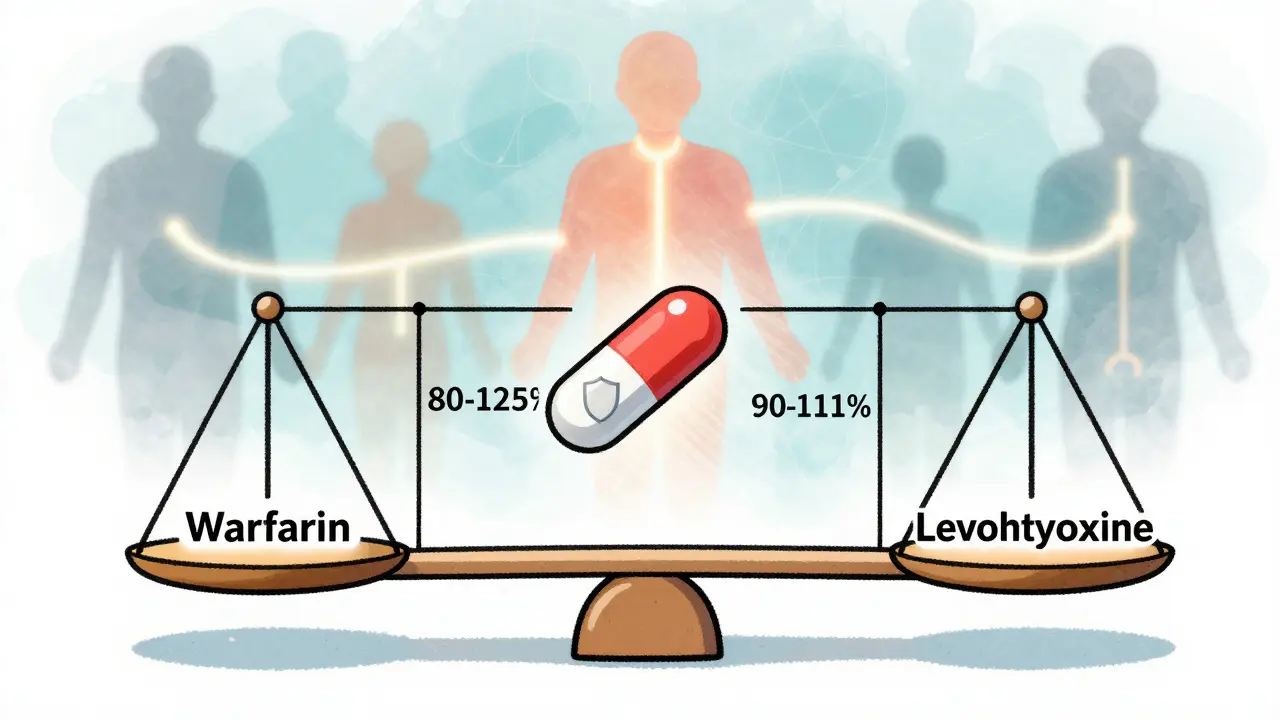
For drugs with a narrow therapeutic index - like warfarin, levothyroxine, or phenytoin - even a 10% difference in exposure can cause serious problems. Too little, and a seizure or blood clot might occur. Too much, and you risk internal bleeding or toxicity. For these drugs, regulators have tightened the range to 90-111%. The FDA issued draft guidance for this in 2022, and the EMA already requires it.
Then there are highly variable drugs. Some drugs are absorbed differently from person to person - even the same person on different days. If a drug has a within-subject coefficient of variation above 30%, the standard 80-125% range doesn’t work. That’s where scaled average bioequivalence (SABE) comes in. Instead of a fixed range, the acceptance limits stretch based on how much the reference drug varies in the body. For example, Cmax for a highly variable drug might be allowed to go as high as 143.19% if the variability justifies it. The EMA has used this since 2010. The FDA now allows it under specific conditions.
These exceptions aren’t loopholes. They’re science-based adjustments. The rule was never meant to be rigid. It was meant to be smart.
How Bioequivalence Studies Are Done
Testing bioequivalence isn’t easy. A typical study involves 24 to 36 healthy volunteers. Each person takes both the generic and brand-name drug, often weeks apart, in a randomized order. Blood samples are taken every 15 to 30 minutes for 24 to 72 hours. Then, labs measure the drug concentration in each sample.
The data gets log-transformed. Statistical software calculates the geometric mean ratio and its 90% confidence interval. Both AUC and Cmax must pass. If one fails, the product is rejected - even if the other passes. It’s not enough to be similar in exposure. You must be similar in speed and total amount.
Studies take 8 to 12 weeks to run. Add 4 to 6 weeks for analysis. For complex drugs - like extended-release pills or inhalers - the process can take over a year and cost $2 million or more. That’s why some companies use bioequivalence waivers. If a drug dissolves in water exactly like the brand-name version under strict testing conditions, regulators may skip the human study entirely. The FDA expanded this program in 2021 for simple oral tablets.
Common Misconceptions and Real-World Impact
Online forums are full of fear. Patients worry that generics are “weaker.” Pharmacists sometimes hesitate to substitute. A 2022 survey found that 28% of neurologists had seen occasional problems with generic anti-seizure drugs. But only 4% of them believed it was because of bioequivalence standards. Most issues came from differences in inactive ingredients - fillers, coatings, or manufacturing processes - not the active drug’s absorption.
The Institute for Safe Medication Practices recorded over 1,200 reports of therapeutic equivalence concerns between 2015 and 2022. Only 17% were linked to bioequivalence. The rest? Packaging confusion, pill shape differences, or patient perception.
Here’s what matters: if a generic passes the 80-125% rule, it has been tested against the same gold standard used for brand-name drugs. The FDA’s Orange Book lists over 13,800 approved generic products. None of them were approved without meeting the rule. And since 1984, when the Hatch-Waxman Act created the modern generic approval pathway, generics have filled 90% of U.S. prescriptions - at just 23% of the cost.
What’s Next for Bioequivalence?
The future is moving beyond one-size-fits-all. Researchers are exploring model-informed bioequivalence - using computer simulations to predict how a drug behaves based on genetics, age, or liver function. The FDA has allocated $15 million for this research through 2027.
For complex products - like nasal sprays, topical creams, or injectable suspensions - traditional bioequivalence studies often fail. That’s why the FDA launched its Complex Generics Initiative in 2018, with $35 million a year to develop new testing methods. The WHO and EMA are following suit.
By 2030, bioequivalence might not just say “this drug works like that one.” It could say, “this drug works like that one in patients with CYP2D6 slow metabolism.” Personalized bioequivalence is coming. But for now, the 80-125% rule still holds the line.
Bottom line: the rule isn’t perfect. But it’s reliable. And for the vast majority of drugs, it’s more than enough.
Is the 80-125% rule the same everywhere in the world?
Yes. The 80-125% rule is used by the U.S. FDA, the European Medicines Agency (EMA), the World Health Organization (WHO), Health Canada, and over 50 other countries. This global alignment allows generic drug manufacturers to seek approval in multiple markets using the same data, reducing costs and speeding up access to affordable medicines.
Does the 80-125% rule mean generic drugs are less potent?
No. The rule has nothing to do with how much active ingredient is in the pill. Generic drugs must contain 95-105% of the labeled amount - the same as brand-name drugs. The 80-125% range refers to how much of the drug reaches your bloodstream (pharmacokinetics), not how much is in the tablet. Two identical pills can have the same active ingredient but different absorption rates due to fillers or coatings - and that’s what the rule tests.
Why is a 90% confidence interval used instead of 95%?
A 90% confidence interval allows for a 5% risk of error at the lower end and a 5% risk at the upper end, totaling 10% overall. This balances statistical rigor with practicality. In bioequivalence, the goal isn’t to prove the drugs are identical - it’s to prove they’re close enough to have the same clinical effect. A 95% CI would be too strict for this purpose, requiring unrealistically large studies.
Can a generic drug fail the 80-125% rule and still be safe?
Regulators say no. If a product’s 90% confidence interval falls outside 80-125% for either AUC or Cmax, it’s not approved. This isn’t about theoretical safety - it’s based on decades of data showing that drugs outside this range are more likely to cause clinical issues. Even if a product seems safe in early trials, regulators won’t risk long-term outcomes on unproven data.
Why do some patients say generics don’t work as well?
In most cases, it’s not bioequivalence. It’s perception, packaging, or formulation differences. A pill that looks or tastes different can make patients feel like it’s less effective. Sometimes, inactive ingredients cause side effects - like a coating that irritates the stomach. Rarely, a bioequivalence study may miss subtle differences in high-variability drugs. But post-market data shows these cases are extremely uncommon. The 80-125% rule has protected millions of patients for over 35 years.
Comments
Spenser Bickett
sooooo... you're telling me that the government lets companies make pills that *might* be 20% weaker or 25% stronger and calls it 'equivalent'? 🤡 i mean, i get the math, but my body ain't a lab rat. i've had generics that made me feel like i got hit by a bus and others that felt like nothing happened. this 'rule' is just corporate propaganda dressed up in stats. they don't care if you feel like crap, as long as the numbers look pretty on a spreadsheet.
February 25, 2026 at 08:17
Natanya Green
OMG YES!! I had this exact experience with my generic thyroid med!! One batch made me feel like a zombie, the next made me feel like I had superpowers!! I swear I was going to call the FDA!! 😭 I just didn't realize it was the *absorption* and not the dose!! This post changed my life!! Thank you!! 🙌❤️
February 25, 2026 at 15:55
Brandice Valentino
I mean, the 80-125% rule is such a *basic* pharmacokinetic principle, but it's hilarious how many people confuse it with dosage. Like, no, sweetheart, the tablet has the same amount of active ingredient-what changes is how your gut decides to play nice with the excipients. It's not magic. It's chemistry. And if you can't handle that, maybe don't swallow pills? 🤷♀️
February 26, 2026 at 00:05
Larry Zerpa
Let’s be clear: this 'rule' is a political compromise masquerading as science. The 90% CI was chosen because it allows more products to pass-not because it's clinically optimal. The FDA has admitted that 10% failure risk is acceptable. That means 1 in 10 generics could theoretically be clinically inferior. And yet, we’re told to trust it. That’s not science. That’s negligence wrapped in a lab coat.
February 27, 2026 at 19:00
Lillian Knezek
this is all a lie. the government and big pharma are in cahoots. they want you dependent on their pills. the 80-125% rule? fake. the real data is buried. i know people who got sick after switching. they vanished from the system. don't trust the system. 🕵️♀️
March 1, 2026 at 07:39
Maranda Najar
The elegance of this regulatory framework is nothing short of poetic. The logarithmic transformation of pharmacokinetic parameters-such a sublime application of mathematical elegance to human physiology! The 90% confidence interval, a delicate dance between statistical rigor and clinical pragmatism… it’s almost romantic. And yet, society reduces it to 'pills are pills.' How tragically reductive.
March 2, 2026 at 09:02
Christopher Brown
Other countries use this? Pathetic. America invented modern medicine. We don’t need some global consensus. If we want better generics, we build them here. Not some EU-WHO compromise. This rule is weak. We should demand 95-105% across the board. Period.
March 3, 2026 at 17:51
Sanjaykumar Rabari
i think this is all very complicated. why not just make the pills the same? why all the math? i dont understand. my uncle in india takes generic and he is fine. maybe this is american problem?
March 4, 2026 at 00:41
Kenzie Goode
I really appreciate how thoughtful this breakdown is. It’s so easy to get caught up in fear or misinformation, but this helps clarify the real science behind it. I’ve had friends panic over generic switches, and now I’ll have a clear way to explain why it’s usually not the drug itself. Thank you for the clarity.
March 4, 2026 at 20:12
Khaya Street
I must say, the regulatory harmonization across jurisdictions is a marvel of international cooperation. It’s rare to see such alignment in public health policy. The 80-125% rule is not merely a technical standard-it is a testament to global scientific consensus. Kudos to the agencies involved.
March 6, 2026 at 15:17
Christina VanOsdol
I just read this and I’m literally crying 😭😭😭 This is the most important thing I’ve learned this year. I’ve been on 5 different generics for my blood pressure and thought I was going crazy. Now I know it’s the coating! Not the drug! I’m telling everyone. This deserves a TED Talk 🙏💖
March 6, 2026 at 15:46
Emily Wolff
The 80-125% rule? It’s a joke. If you need a 20% buffer to prove equivalence, the standard is broken. Real medicine doesn’t work in approximations.
March 7, 2026 at 01:03
Lou Suito
You say the rule is reliable. But you also say it’s not perfect. So which is it? If it’s not perfect, why do we trust it? Hypocrisy wrapped in data.
March 8, 2026 at 02:12
Cory L
This is why I love science. It’s not about being perfect-it’s about being *practical*. The 80-125% rule lets millions get life-saving meds for $4 instead of $400. Yeah, there are edge cases. But the system works better than it ever has. Let’s not throw out the baby with the bathwater.
March 8, 2026 at 21:53